
-
解析 FDA 工廠審查:應對策略與專業指導
一、FDA 工廠審查全貌FDA 作為美國健康與人類服務部(DHHS)的重要分支,肩負著監管全國藥品、食品、生物制品、化妝品、獸藥、醫療器械及診斷用品等的重任。對于醫療器械生產企業而言,QSR820(21 CFR 820)是必須嚴格遵守的質量管理體系法規。該法規依據產品風險等級,對不同類別的醫療器械生產企業進行抽查,以確保企業符合 FDA 的相關法規要求。一般而言,一類產品約 4 年抽查一次,二、三類產品約 3 年抽查一次。然而,若企業產品在海關出現問題或在美國發生不良事件,抽查頻次則會相應增加。2024 年 1 月 31 日,FDA 發布了《質量管理體系法規(QMSR)最終規則》,對《質量體系法規(QSR)》(21 CFR 820)中的器械現行良好生產規范(CGMP)要求進行了修訂,納入了國···
307

-
誠邀各位撥冗蒞臨第91屆CMEF
CMEF(全稱:中國國際醫療器械博覽會)始創于1979年,展會歷經40余年的積累和沉淀,現已發展成為亞太地區集醫療器械全產業鏈、集產品技術、新品首發、采購貿易、品牌傳播、科研合作、學術論壇、教育培訓為一體的醫療器械博覽會,旨在助力醫療器械行業的健康快速發展。 展會覆蓋了醫療器械全產業鏈、集產品技術、新品首發、采購貿易、品牌傳播、科研合作、學術論壇、教育培訓,是國際領先的全球化綜合服務平臺。 本次瑞恩尼及其旗下廣州助醫通聯袂于8號館8.1C07展位,歡迎各位撥冗蒞臨展位交流、指導、合作洽談!!
158
-
快速通過CE認證的秘密通道!
在當今全球化的商業舞臺上,企業的產品和服務不僅要滿足國內市場的需求,更要跨越國界,贏得國際市場的認可。歐盟作為一個龐大的經濟體,對產品質量、安全性和環保標準有著嚴格的要求。這就引出了一個重要的認證——CE認證。作為專業的CE認證咨詢顧問,我深知這一認證對于企業意味著什么,以及如何幫助它們順利獲得這一“通行證”。CE認證,全稱為Conformité Européenne,是制造商宣告其產品符合歐盟健康、安全和環境保護要求的一種自我聲明。它不僅僅是一個標志,更是企業進入歐洲市場的敲門磚。通過CE認證的產品,意味著它們已經通過了嚴格的測試和評估,可以在歐洲經濟區(EEA)自由流通。對于許多中國企業來說,CE認證不再是可選項,而是必答題。隨著中歐貿易的蓬勃發展,越來越多的中國產品涌向歐洲市場。但要想···
104
-
FDA工廠檢查,品質背后的真相揭秘!
在當今社會,健康與醫療安全已經成為了公眾關注的焦點。在這個背景下,FDA(美國食品藥品監督管理局)對制藥工廠的檢查顯得尤為重要。這些檢查不僅是對藥品生產過程的一種監督,更是確保藥品安全性和有效性的重要手段。FDA工廠檢查的核心目的在于驗證藥品生產是否遵循了既定的質量標準和法規要求。這一過程包括對生產設備、原材料、生產工藝、質量控制以及員工培訓等各個方面的審查。通過這些嚴格的檢查,FDA能夠確保市面上流通的藥品是安全且有效的,從而保護消費者的健康不受損害。隨著科技的進步和人們對健康意識的提高,FDA的檢查標準也在不斷地提升。這意味著制藥企業必須投入更多的資源和精力來滿足日益嚴格的監管要求。例如,為了確保數據的透明性和可追溯性,許多制藥公司已經開始采用先進的信息技術系統來管理其生產和供應鏈流程。···
100
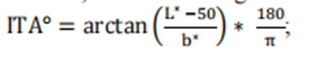
-
醫用脈搏血氧儀 - 非臨床和臨床性能測試、標簽和上市前提交建議)》指南草案
2025版Pulse Oximeters for Medical Purposes - Non-Clinical and Clinical Performance Testing, Labeling, and Premarket Submission Recommendations指南草案與2013版Pulse Oximeters – Premarket Notification Submissions [510(k)s] Guidance for Industry and Food and Drug Administration Staff指南的差異與主要變化點2025年初,FDA發布了《Pulse Oximeters for Medical Purposes - Non-Clinical···
126
-
MDSAP認證如何助力產品快速出海?
在全球醫療器械市場中,合規性和安全性始終是企業立足的基石。隨著國際貿易的不斷發展,醫療器械單一審核程序(MDSAP)認證成為了許多醫療器械制造商拓展國際市場的關鍵。然而,面對復雜多變的國際法規和嚴格的監管要求,如何高效地通過MDSAP認證,確保產品在多個國家市場的順利流通,成了眾多企業亟待解決的問題。MDSAP認證是一個旨在簡化醫療器械進入加拿大、澳大利亞、巴西以及日本市場過程的程序。它允許參與該計劃的國家的監管機構接受彼此的審核結果。這意味著如果一個產品通過了其中一個國家的審核,它將自動被其他參與國家接受,從而大大減少了重復工作,提高了市場準入效率。要成功獲得MDSAP認證,首先需要對目標市場的法規有深入的了解。每個國家都有其特定的醫療器械法規和標準,比如加拿大的健康加拿大、澳大利亞的治療···
110

