
-
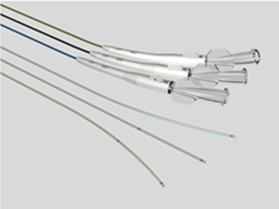
微導管NMPA注冊要點
微導管NMPA注冊要點一、微導管的簡介微導管通常由導管管身、不透射線標記、接頭等結構組成。管身通常較細且柔軟,表面可帶有親水涂層。用于向血管系統中注入診斷試劑(如造影劑)、治療試劑(如藥物制劑、栓塞材料)和適當的器械(如支架、彈簧圈)等。微導管在冠狀動脈粥樣硬化性心臟病(冠心病)的介入治療領域,特別是在應對極具挑戰性的慢性閉塞病變(CTO)時,扮演著不可或缺且至關重要的角色。隨著全球心血管疾病發病率的持續攀升,以及患者對微創手術安全性、有效性追求的日益增長,先進的導管技術成為了推動醫療進步的關鍵力量。微導管,憑借其超細的尺寸、卓越的靈活性和精確的操控性,完美契合了復雜冠脈介入手術的需求。它們能夠輕松穿越蜿蜒曲折、狹窄甚至閉塞的血管通道,為醫生提供了前所未有的治療路徑,實現了對病變部位的精準導···
404
-
NMPA注冊的流程步驟
在中國,醫療器械的注冊是確保產品安全、有效并符合國家標準的重要環節。這一過程由國家藥品監督管理局(National Medical Products Administration, NMPA)負責監管和執行。NMPA注冊的流程相對復雜,涵蓋了多個階段和詳細步驟,以確保申請產品的質量和合規性。以下是NMPA注冊流程的一般步驟概述:一、準備階段1. 收集資料與文件準備在申請NMPA注冊之前,申請人需要收集和準備一系列與產品相關的文件和資料。這些文件包括但不限于:企業基本信息:包括公司營業執照、生產許可證等。產品技術文件:產品的技術規格、設計圖紙、制造工藝、材料成分等詳細說明。質量管理體系文件:證明企業生產過程符合質量管理體系要求的文件,如ISO 13485質量管理體系認證證書。臨床試驗數據(如適···
261
-
MDSAP認證五個參與國家的審核要求
MDSAP(Medical Device Single Audit Program)認證是一個旨在通過單一審核程序,簡化并統一醫療器械制造商進入五個主要國際市場(澳大利亞、巴西、加拿大、日本和美國)的復雜過程。這一認證計劃不僅提高了審核效率,還確保了醫療器械在全球范圍內的質量和安全性。以下是MDSAP認證中五個參與國家審核要求的詳細闡述。一、美國(FDA)的審核要求美國的食品藥品監督管理局(FDA)對醫療器械的監管尤為嚴格,其MDSAP審核要求涵蓋了質量管理體系的全面評估。制造商需確保其質量管理體系符合ISO 13485標準,并額外滿足FDA關于醫療器械注冊、許可、上市后監督和不良事件報告的具體法規。FDA特別強調風險管理的重要性,要求制造商建立并實施有效的風險管理體系,對產品從設計、開發到···
326
-
GMP注冊咨詢服務包括哪些
在醫藥行業中,GMP(Good Manufacturing Practices,良好生產規范)不僅是藥品生產質量管理的基石,也是藥品進入國際市場的重要門檻。GMP注冊咨詢服務作為連接藥品生產企業與監管機構之間的橋梁,其重要性不言而喻。本文將深入探討GMP注冊咨詢服務所涵蓋的關鍵內容,幫助企業更好地理解并高效推進這一過程。一、GMP法規咨詢GMP注冊咨詢服務的首要任務是提供全面而準確的GMP法規咨詢。這包括但不限于國際GMP標準(如歐盟GMP、FDA cGMP)及各國特定法規要求的解讀。咨詢專家會詳細分析企業當前的生產管理體系與GMP標準的差距,識別潛在的風險點,并提出針對性的改進建議。此外,隨著法規的不斷更新,咨詢服務還需保持對最新法規動態的跟蹤,確保企業能夠及時調整策略,滿足最新的合規要求···
265
-
FDA工廠檢查應提前準備什么材料
在醫藥及食品行業中,FDA(美國食品藥品監督管理局)的工廠檢查是確保產品安全、質量和合規性的重要環節。對于任何即將接受FDA檢查的企業而言,充分而周密的準備是至關重要的。這不僅關乎企業的聲譽,更直接影響到產品的市場準入及未來的發展前景。以下是一份詳盡的指南,概述了企業在FDA工廠檢查前應提前準備的關鍵材料,以確保檢查的順利進行。一、企業基本信息與資質文件首先,企業應確保所有基本的注冊、許可和認證文件齊全且更新至最新狀態。這包括但不限于:企業注冊證書:FDA注冊證明,確認企業已在美國FDA注冊并獲得唯一設施識別碼(FEI)。生產許可證:國內或國際相關監管機構頒發的生產許可證書,證明企業具備合法生產的資質。GMP(良好生產規范)認證:對于藥品生產企業,需提供GMP認證證書及其最新審核報告,展示企···
303
-
NMPA注冊監督檢查和持續合規
在中國,醫療器械的市場準入和持續合規管理是一項至關重要的工作,直接關系到公眾的健康與安全。國家藥品監督管理局(NMPA)作為醫療器械監管的主要機構,承擔著確保醫療器械質量、安全性和有效性的重要職責。NMPA注冊監督檢查和持續合規不僅是對醫療器械制造商的嚴格要求,也是保障醫療市場健康發展的重要手段。一、NMPA注冊監督檢查的重要性NMPA注冊監督檢查是醫療器械進入市場前的一道重要防線。這一過程涉及對醫療器械制造商提交的技術文件、質量管理體系、臨床試驗數據(如適用)等多方面的詳細審核。技術評審是其中的核心環節,旨在評估醫療器械的技術規格、設計、性能和制造過程是否符合中國的法規和技術標準。此外,對于高風險或特定類別的醫療器械,NMPA還會進行現場審核,以驗證生產設施、質量管理體系和產品制造過程是否···
255

